
QSit out – In with the New FDA Inspection Program!

For those in the medical device industry, you’ve probably heard the buzz: on 01/30/2026, the Food and Drug Administration (FDA) reminded the industry of the implementation of the Quality Management System Regulation (QMSR) on 02/02/2026 AND that they would no longer be using the long-standing Quality System Inspection Technique (QSIT) for inspecting facilities. Instead, they’ve rolled out and replaced QSIT with the updated – Inspection of Medical Device Manufacturers Compliance Program(CP 7382.850) [1] . What does this mean for medical device manufacturers? Let’s QSIT down and take a deeper look!
As of February 2, 2026, the FDA will no longer use the following documents: Inspection of Medical Device Manufacturers(7382.845) and Medical Device PMA Preapproval and PMA Postmarket Inspections (7383.001). The updated program aligns with Quality Management System Regulation (QMSR) requirements, describes the QMSR inspection process, and updates regulatory procedures and program contacts [2] .
The Previous Inspection Approach: QSIT
For years, QSIT was the FDA’s go-to method for inspecting a medical device manufacturer’s Quality Management System (QMS). QSIT had a set structure focusing on four main areas: management controls, design controls, product and process controls, and corrective and preventive actions. Inspections with QSIT often felt predictable because companies knew the main topics inspectors would cover.
The New Approach: CP 7382.850
FDA’s upgraded method, (CP7382.850), takes a more risk-based and flexible approach. Instead of following a set checklist, inspectors will focus on areas that are most likely to impact patient and user safety using reviews of past issues, complaints, recalls, and emerging risks in a company’s device category to make an assessment. This means every inspection could look a little different depending on your company’s history and the specific devices made.

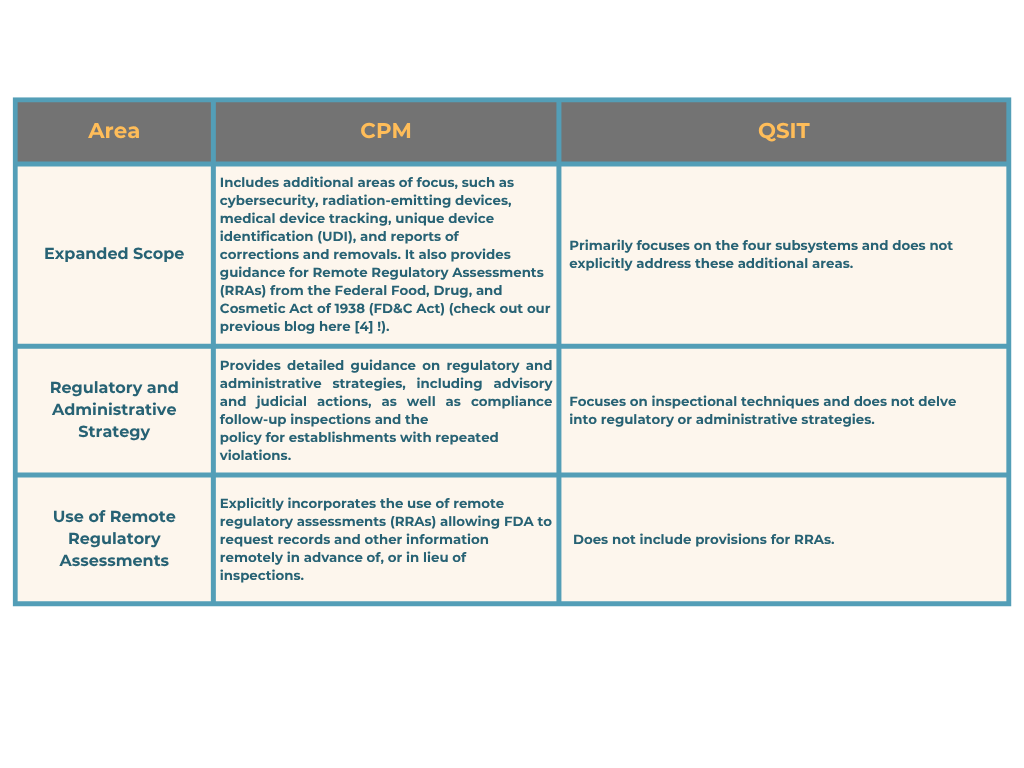
The following are QMS areas that are interconnected and evaluated during FDA inspections to ensure compliance with the QMSR and to identify risks that could adversely impact patients and users (CP 7382.850):
1. Management Oversight:
Focuses on ensuring that top management plans, establishes, and maintains an effective QMS.
Includes management commitment, quality policy, quality objectives, management review, provision of resources, and risk-based decision making.
2. Design and Development
Ensures that design and development activities result in safe and effective medical devices that meet their intended use.
Includes elements such as design planning, inputs, outputs, review, verification, validation, transfer, and control of design changes.
3. Production and Service Provision
Ensures that production and service provision processes are planned, monitored, and controlled to produce safe and effective medical devices.
Includes elements such as infrastructure, work environment, contamination control, production controls, sterilization processes, traceability, and preservation of product.
4. Change Control
Ensures that changes to products, processes, and systems are adequately evaluated for risk and impact before implementation.
Includes elements such as QMS changes, software changes, product and process changes, and purchasing changes.
5. Measurement, Analysis, and Improvement
Ensures that monitoring, measurement, analysis, and improvement activities are effective in identifying and reducing risks that impact the product and/or the QMS.
Includes elements such as complaint handling, feedback, internal audits, data analysis, corrective actions, preventive actions, and control of nonconforming products.
6. Outsourcing and Purchasing
Ensures that outsourced processes, activities, and purchased products are effectively monitored and controlled to ensure product conformity.
Includes elements such as outsourcing, purchasing processes, and control of purchased products.
While QSIT provided a structured approach to inspecting medical device manufacturers, the Compliance Program Manual for Program 7382.850 is more dynamic and tailored to real-world risks. Establishments must now switch their mindsets from “passing a checklist” to “actively managing risks” in preparation for their next inspection, and more importantly, keeping patients and users safe.
Rest in peace QSIT (1999-2026).
Thanks for stopping by, keep checking back as we follow industry trends!
Have questions? Contact us here!
References