
Back to Basics: Records and Documents

Welcome back! Today we’re revisiting two areas required by the Food and Drug Administration (FDA) for regulated manufacturing environments in the human cells, tissues, and cellular and tissue-based products (HCT/Ps), medical devices, and drug industries. In an FDA-regulated manufacturing environment, two words come up constantly: “Records” and “Documents”. They sound similar but they’re not the same and mixing them up or omitting one or the other can create real compliance problems in your quality system. FDA regulations expect companies to control both. Today we’re taking it back to the basics, let’s walk through some details and risks depending on which regulation applies.
What’s the difference between a document and a record? In plain terms, a document tells what companies are supposed to do, while a record proves what was actually done. A document is an instruction such as standard operating procedures (SOPs), work instructions, forms, specifications, templates, methods, and quality manuals. These items tell employees how work should be done, since this is direct work, it must be reviewed, approved, updated when applicable, and protected from accidental or unauthorized changes.
A record, on the other hand, is the evidence left behind after work happens. Examples include completed batch records, donor eligibility records, test results, equipment logs, training records, complaint files, deviation investigations, and release decisions. Records are proof and they show what happened, when it happened, who did it, and whether the company followed its own procedures and regulatory requirements.
Here’s a quick breakdown of how different regulations approach records and documents in our industry of regenerative medicine and cellular therapeutics.
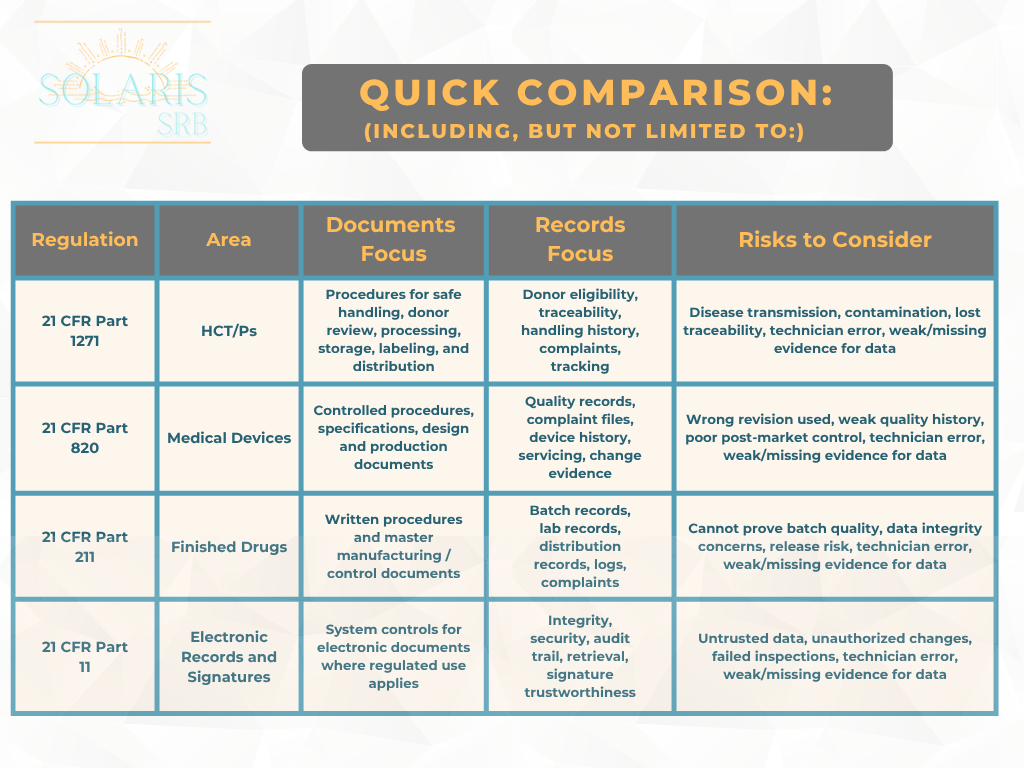
21 CFR Part 1271 Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps) [1]
Purpose: Prevent communicable disease transmission in human cells and tissues.
Documents focus: Written procedures for donor screening, testing, processing, storage, labeling, and distribution.
Records focus: Donor eligibility, handling history, complaints, tracking, and traceability.
21 CFR Part 820 Medical Devices [2]
Purpose: Control the full device quality management system across the product life cycle.
Documents focus: Controlled procedures, specifications, design documents, and production instructions.
Records focus: Complaint files, device history, device master records, servicing, and quality system evidence.
21 CFR Part 211 Finished Drugs [3]
Purpose: Ensure finished drugs are consistently manufactured and controlled.
Documents focus: Written procedures and master production/control records.
Records focus: Batch records, lab records, distribution records, equipment logs, and complaints
21 CFR Part 11 Electronic Records and Signatures [4]
Purpose: Set rules for electronic records and electronic signatures.
Documents focus: Electronic systems must be validated, secure, and access controlled.
Records focus:Audit trails, record protection, reliable retrieval, and signature worthiness.
No matter which FDA regulation applies, establishments should keep this basic rule in mind: say what you do, do what you say, and keep proof that you did it. The exact emphasis changes by industry. Tissue establishments must protect donor and recipient safety through traceability and communicable disease controls. Device manufacturers must maintain a robust quality system across the product life cycle. Drug manufacturers must preserve a complete and reliable manufacturing detail for each batch. Any company using electronic systems for regulated records must ensure those systems are validated, secure, and inspection ready. Documentation problems are rarely just paperwork problems. In FDA regulated industries, poor control of documents and records lead to product quality issues, patient safety risks, recalls, warning letters, and loss of business trust. Records and documents are not just administrative details, they are key parts of an establishmented quality system.
Questions? Contact us here!
Thanks for joining, check back next time as we keep up with industry trends!
Reference
[1] https://www.ecfr.gov/current/title-21/chapter-I/subchapter-L/part-1271
[2] https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820
[3] https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211
[4] https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-11